



甲基丙二酸血症(methylmalonic acidemia, MMA)是一种常染色体隐性遗传的代谢性疾病,由于缺乏甲基丙二酰辅酶A变位酶(methylmalonyl-CoA mutase,MCM)及其辅因子维生素B12,导致甲基丙二酸等中间代谢产物在体内堆积的一类有机酸血症[1]。该疾病在临床上较罕见,据统计全球MMA的发病率1:25×104~1:4.8×104[2]。按起病年龄可分为早发型(≤1岁)和迟发型(>1岁)两种类型,迟发型可在任何年龄出现,起病隐匿,临床表现复杂多样无特异性,漏诊、误诊率较高,可致多系统器官受损[3]。本文回顾性分析1例以情感障碍为首发症状的迟发型MMA患者的诊疗过程,并复习相关文献,帮助提高临床医生对该疾病的认识,以便及早识别该疾病,减少病人的痛苦,改善预后。
1 病例资料患者女性,27岁,既往患有“双相情感障碍”10年,近1年未治疗,表现基本正常。本次因“兴奋、话多、行为紊乱1周”于2018年6月2日入济宁市精神病防治院精神科住院治疗。
现病史:患者入院前1周自觉高兴,精力旺盛,认为自己能力强、本事大,到处逛街,乱花钱,穿衣打扮花哨,有时举止轻浮,爱管闲事,家人劝阻时发脾气扔东西,在家难以管理。睡眠需求明显减少,饮食、二便均正常。
患者住院第10天,表现情绪稳定,言语活动减少,不再吹嘘,并且白天睡眠增多,第15天患者开始变得接触被动,表情呆板,思维联想迟缓,生活被动懒散,个人卫生不知料理,意志活动减退,整日趴在桌子上睡觉,有时诉下肢无力发软,走路不稳,考虑患者转为抑郁相。第24日发现患者尿裤子,症状仍以精神抑制为主,颅脑MRI显示脑沟裂及室池较宽,请神经内科医师会诊,查体无阳性体征。第30天患者开始出现大小便失禁,噎食,动作笨拙,行走困难,问话多不答,生活难以自理,查体见患者口周皮肤有疱疹,余无异常。考虑可能为器质性病变,由于本院医疗水平有限,建议到上级综合医院进一步诊疗,于2018年7月13日转诊于山东省立医院住院治疗。
既往史:患“双相情感障碍”10年,近1年来未服药治疗,表现正常。否认有肝炎、结核等传染病史,否认有冠心病、高血压等病史,预防接种随当地,无食物、药物过敏史。
个人史:母孕期正常,足月顺产,自幼发育良好,适龄上学,成绩一般,初中文化。务农,劳动能力可。无烟酒等不良嗜好,无精神活性物质使用史。
婚育史:月经规律,无痛经史,末次月经不详。已婚,夫妻关系良好,育1子。配偶及孩子身体健康。
家族史:否认两系三代中有精神异常者。否认其他遗传病。
体格检查:2018年6月2日体格检查无明显异常。2018年7月13日神经系统检查:患者平卧位,神志不清,烦躁,言语模糊。颈项强直,双侧瞳孔等大等圆,d=3.5mm,双侧对光反射灵敏,眼球动度可,无震颤。四肢肌力5级,肌张力(
精神检查:意识清,接触主动,态度热情,检查、治疗及护理合作,生活自理能力可,无时间、地点、人物定向力障碍,无自我意识障碍。未引出明确的感知觉异常及感知障碍综合征。思维奔逸,自觉聪明,脑子灵活。未查妄想及其他病理性信念。情感高涨,言语活动增多,精力旺盛,注意力随境转移,记忆力、智能一般。意志活动病理性增强,否认有病,无自知力。
2 辅助检查1) 躁狂量表(BRMS)测查结果:总分22分,表明存在严重躁狂症状。
2) 实验室检查结果:6月3日查血尿常规、肝功、肾功、心肌酶、血糖等均无异常,梅毒螺旋体特异抗体、乙肝五项等均阴性。7月8日复查血常规、肝肾功能、血糖等无异常,心肌酶结果示CK886U/L、LDH286U/L、HBDH213U/L较正常值高。7月15日尿常规+沉渣示尿糖1(+),尿酮体2(+),尿蛋白1(+),尿白细胞升高;风疹病毒IgG抗体30.6IU/ml,巨细胞病毒IgG抗体87U/ml,单纯疱疹病毒IgG抗体23.2COI、IgM抗体1.32COI,细小病毒B19 IgG抗体4.3IU/ml,均高于正常值;ASO488KU/L(偏高); 血同型半胱氨酸215.6μmol/L,β-羟丁酸290.68mmol/L(均偏高)。7月16日细胞免疫功能检查提示CD3+49.88%,CD3+CD4+26.88%(均偏低), CD19+27.28%(偏高),女性肿瘤标志物、凝血功能、铜蓝蛋白、叶酸、维生素B12、PCT等均无异常。7月26日血有机酸结果提示丙酰基肉碱显著升高。
3) 电生理检查结果:6月3日常规脑电图示正常范围脑电图。7月11日脑电图提示低波幅β型脑电图。心电图无明显异常。
4) 影像学检查:6月21日颅脑CT示脑沟裂及室池较宽,脑实质未见异常信号影,中线结构居中。7月10日颅脑MRI示小脑多发缺血灶、海马萎缩。7月11日颅脑MR提示双侧小脑半球异常信号,考虑炎症,变性或中毒性疾病待排。7月16日颅脑+胸椎MR提示小脑弥漫性异常信号(DWI)异常高信号,病变范围无变化,符合脑炎MRI表现,不排除缺血性脑改变;脑血管远端分支减少MRA表现,痉挛?符合双侧海马体积小或萎缩MRI表现;胸椎MRI未见异常。
5) 脑脊液检查:脑脊液无色透明,压力90mmH2O, 潘氏试验阴性,葡萄糖、蛋白含量均无明显异常,脑脊液病原体二代测序、自免脑抗体、中枢神经系统脱髓鞘系列抗体均阴性。
诊疗过程:首诊医生根据患者的临床表现及相关辅助检查结果,诊断为双相情感障碍,不伴精神病症状的躁狂发作,给予情感稳定剂丙戊酸钠等,后患者出现精神抑制,合并舍曲林治疗,但患者病情无明显缓解,反而出现行走不稳,二便失禁,意识模糊,并进行性加重。考虑患者存在器质性病变,建议转诊至省级医院神经内科。由于患者存在病毒感染征象,综合体格检查、实验室检查、影像学检查等,上级综合医院医师初步诊断为病毒性脑炎?但未排除自身免疫性脑病、MMA的诊断,后行腰椎穿刺术检验脑脊液,血有机酸及基因检测(基因检测结果家属不能提供)确诊为1.MMA;2.单纯疱疹病毒感染。治疗上停用精神科用药,给予更昔洛韦注射液250mg q12h、甲泼尼龙注射液500mg qd等药物静滴,甲钴胺注射液1mg qd静脉注射,口服叶酸、维生素B6、甲钴胺、左旋肉碱、甜菜碱等药物,营养支持治疗,于7月27日好转出院,出院时意识清,反应可,言语清晰,可坐起,四肢肌力正常,肌张力(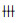
鉴别诊断:1)病毒性脑炎。患者可表现为精神行为异常,严重者可影响智力,但该病发病前多有前驱感染史,起病较急,伴体温升高,部分患者可见口唇疱疹。本例患者脑脊液生化检查正常,脑脊液病原学检查为阴性,故可排除该诊断。2)自身免疫性脑炎。患者通常亚急性起病, 病情发展迅速,多短于3个月,并可出现意识状态改变,性格改变、精神症状等,影像学检查可见脱髓鞘病变和炎性病变。本例患者起病快,存在兴奋、行为紊乱等精神症状,病情加重后有意识障碍,颅脑磁共振显示小脑炎性病变,应与该病鉴别,但本例病人脑脊液自免脑抗体、中枢神经系统脱髓鞘系列抗体均阴性,故可排除。3)桥本脑病。本病多急性或亚急性起病,中年女性多见,临床可分为卒中样发作型和持续进展型,后者多表现为精神症状如幻觉、妄想、激越、易怒,抑郁、淡漠、行为异常等。本例患者精神症状丰富,起病快,应与该病鉴别,但该病人甲状腺功能正常,脑脊液生化检查正常,伴有机酸水平升高,因此可排除。
3 讨论MMA是有机酸代谢病中最常见的类型[4]。当MCM及辅酶B12缺乏时体内氨基酸、脂肪酸等的代谢途径受损,丙酸、甲基丙二酸等中间毒性代谢产物在体内蓄积,三羧酸循环受限,线粒体能量合成障碍,导致神经系统、血液系统、免疫系统等多系统功能障碍[5]。依据发病机制可分与MCM缺陷相关的mut型和腺苷钴胺合成缺陷相关的cbl型[6],包括mut0(完全无活性型)、mut-(部分残余活性型)、cblA、cblB、cblC、cblD、cblF、cblH等亚型。其中mut型及cbl型中的A、B、H亚型患者仅表现MMA,而cblC,cblD和cblF型则合并同型半胱氨酸血症[7]。在我国cblC型是最常见的合并同型半胱氨酸血症的MMA[8]。本文中的病例虽没有提供基因型检测结果,但该患者同时伴有同型半胱氨酸的升高,推测是cblC型的可能性较大。
MMA因其主要好发于婴幼儿时期,通常被认为是一种婴幼儿代谢异常性疾病,成人期起病较为罕见,目前国内外已有一些成人起病的病例报告[1-3, 8]。MMA诊断和管理指南提出该疾病可在任何年龄出现急性或慢性症状,急性症状可表现为呕吐和喂养困难等胃肠功能的紊乱,嗜睡、惊厥甚至昏迷等神经系统症状,呼吸窘迫、过度通气等呼吸系统问题,以及血液系统、免疫系统、肾脏损害等。慢性症状可表现为肌张力障碍、发育迟缓(包括学习障碍,智力残疾)、运动障碍、精神症状(幻觉,精神病发作)等[1]。研究表明在迟发型病例中,神经系统的损伤往往比较明显,临床神经影像学显示患者出现基底神经节(尤其苍白球)对称变性,脑萎缩,脑白质脱髓鞘病变,脑水肿等,本例患者神经系统的功能改变及影像学表现均符合相关文献的报道,分析这些改变与甲基丙二酸、丙酸、同型半胱氨酸等中间毒性产物的神经毒性以及脑能量供给不足有关[8-10]。既往文献报道仅有极少数病例出现精神症状,几乎无躁狂或抑郁发作,Wu等[2]首次报道了两例以躁狂或抑郁为首发症状的MMA并分析了其基因型属于cblC型,可见情感性障碍可能是迟发性MMA的首发症状之一,对此本文的报道又是一个有力的证据。
由于MMA临床表现复杂多样,个体差异性大,易被误诊、漏诊。本例患者在诊疗过程中曾被诊断为双相情感障碍,病毒性脑炎,给情感稳定剂、抗病毒治疗后病情仍无好转,最后通过检测血有机酸显示丙酰基肉碱显著升高确诊为MMA。因此, 当临床上遇到原因未明的神经精神症状时,应考虑到是否为MMA,可通过气相色谱-质谱(GC-MS)法检测血、尿、脑脊液有机酸水平明确诊断,必要时可做基因检测明确分型[11]。本例患者经维生素B族药物治疗效果良好,分析属于维生素B12反应型,对此类患者推荐使用甲钴胺、腺苷钴胺、左旋肉碱、叶酸联合治疗,对伴有高同型半胱氨酸血症的患者加用甜菜碱,另外要限制患者饮食蛋白的摄入量[12-13]。相较于早发型、维生素B12无反应型患者,本例这种维生素B12反应型的迟发型患者生存率较高,预后较好[14]。
| [1] |
Baumgartner MR, Hörster F, Dionisi-Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia[J]. Orphanet J Rare Dis, 2014, 9: 130. DOI:10.1186/s13023-014-0130-8 |
| [2] |
Wu LY, An H, Liu J, et al. Manic-depressive psychosis as the initial symptom in adult siblings with late-onset combined methylmalonic aciduria and homocystinemia, cobalamin C type[J]. Chin Med J (Engl), 2017, 130(4): 492-494. DOI:10.4103/0366-6999.199826 |
| [3] |
陈文琳, 李鑫, 毛翛, 等. 晚发型甲基丙二酸尿症合并同型半胱氨酸血症临床及分子遗传学特点研究[J]. 国际神经病学神经外科学杂志, 2017, 44(2): 132-138. DOI:10.16636/j.cnki.jinn.2017.02.005 |
| [4] |
Deodato F, Boenzi S, Santorelli FM, et al. Methylmalonic and propionic aciduria[J]. Am J Med Genet C Semin Med Genet, 2006, 142C(2): 104-112. DOI:10.1002/ajmg.c.30090 |
| [5] |
蒋雯巍, 蒋雨平. 甲钴胺代谢及其相关性疾病[J]. 中国临床神经科学, 2010, 18(2): 203-207, 212. DOI:10.3969/j.issn.1008-0678.2010.02.018 |
| [6] |
Martínez MA, Rincón A, Desviat LR, et al. Genetic analysis of three genes causing isolated methylmalonic acidemia:identification of 21 novel allelic variants[J]. Mol Genet Metab, 2005, 84(4): 317-325. DOI:10.1016/j.ymgme.2004.11.011 |
| [7] |
Wang F, Han L, Ye J, et al. Analysis of the MUT gene mutations in patients with methylmalonic acidemia[J]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 2009, 26(5): 485-489. DOI:10.3760/cma.j.issn.1003-9406.2009.05.001 |
| [8] |
Wang X, Sun W, Yang Y, et al. A clinical and gene analysis of late-onset combined methylmalonic aciduria and homocystinuria, cblC type, in China[J]. J Neurol Sci, 2012, 318(1-2): 155-159. DOI:10.1016/j.jns.2012.04.012 |
| [9] |
Okun JG, Hörster F, Farkas LM, et al. Neurodegeneration in methylmalonic aciduria involves inhibition of complex Ⅱ and the tricarboxylic acid cycle, and synergistically acting excitotoxicity[J]. J Biol Chem, 2002, 277(17): 14674-14680. DOI:10.1074/jbc.M200997200 |
| [10] |
Wajner M, Coelho JC. Neurological dysfunction in methylmalonic acidaemia is probably related to the inhibitory effect of methylmalonate on brain energy production[J]. J Inherit Metab Dis, 1997, 20(6): 761-768. DOI:10.1023/A:1005359416197 |
| [11] |
Fowler B, Leonard JV, Baumgartner MR. Causes of and diagnostic approach to methylmalonic acidurias[J]. J Inherit Metab Dis, 2008, 31(3): 350-360. DOI:10.1007/s10545-008-0839-4 |
| [12] |
Martinelli D, Deodato F, Dionisi-Vici C. Cobalamin C defect:natural history, pathophysiology, and treatment[J]. J Inherit Metab Dis, 2011, 34(1): 127-135. DOI:10.1007/s10545-010-9161-z |
| [13] |
Thakkar K, Billa G. Treatment of vitamin B12 deficiency-methylcobalamine cyancobalamine hydroxocobalamin-clearing the confusion[J]. Eur J Clin Nutr, 2015, 69(1): 1-2. DOI:10.1038/ejcn.2014.165 |
| [14] |
Fei W, 韩连书. 甲基丙二酸血症诊治研究进展[J]. 临床儿科杂志, 2008, 26(8): 724-727. DOI:10.3969/j.issn.1000-3606.2008.08.023 |